Getting Started: mapping scRNA-seq to bulk RNA-seq atlas
Jiadong Mao
2025-12-02
getting_started.RmdHighlights
Bulk reference + single-cell query;
Learning two layers of phenotypes: cell types and sample source;
Fully data driven gene selection;
Visualising transitional cell states in phenotype space.
Introduction
PhiSpace is inspired by stem cell research, where the focus is often on transitional cell states instead of terminally differentiated cell types.
In the first case study in our manuscript, we used
- Reference bulk RNA-seq: Stemformatics DC atlas
- Query scRNA-seq: Rosa et al. (2022)
Dendritic cells (DCs) are a type of immune cells. DCs are relatively rare in human blood samples. Hence it is desirable to culture in vivo like DCs using in vitro methods. Rosa et al. (2022) claimed that they successfully reprogrammed human embryonic fibroblasts (HEFs) into induced DCs after 9 days in vitro cell culturing.
The Stemformatics DC atlas is a bulk RNA-seq atlas of different subtypes of human DC samples (FACs sorted). Moreover, these DC samples had different sample sources, including in vitro, in vivo, ex vivo, etc. Hence the DC atlas is comprehensive enough for veriying the cell identity of induced DCs from Rosa et al. (2022).
Load the packages.
# Make sure you've installed the latest version of PhiSpace
# BiocManager::install('jiadongm/PhiSpace/pkg')
suppressPackageStartupMessages(library(PhiSpace))
# Tidyverse packages
suppressPackageStartupMessages(library(ggplot2))## Warning: package 'ggplot2' was built under R version 4.5.2
suppressPackageStartupMessages(library(dplyr))
suppressPackageStartupMessages(library(magrittr))
suppressPackageStartupMessages(library(ggpubr))
suppressPackageStartupMessages(library(tidyr))
suppressPackageStartupMessages(library(qs)) # For fast saving and writing R objects
# Other utils
suppressPackageStartupMessages(library(ComplexHeatmap)) # plot heatmap
suppressPackageStartupMessages(library(zeallot)) # use operator %<-%
suppressPackageStartupMessages(library(plotly)) # plot 3d interative plotsData preparation
Download the processed reference dataset ref_dc.qs and the processed and query dataset query_Rosa_sub.qs.
We first donwsample query data to ensure that we can run this example
on a local machine, otherwise a larger RAM is needed. The function we
use for subsampling is PhiSpace::subsample, which has the
following features
- It allows stratified subsampling, eg subsample a proportion from each cell type;
- When doing stratified subsampling, we can set a minimal number of
cells
minCellNumfrom each cell type; - It sets a random seed implicitly.
The second feature above is to prevent rare cell types from being underrepresented after subsampling. The third feature comes handy as we sometime forget to set seed, but setting random seed is absolutely essential for reproducibility.
dat_dir <- "~/Dropbox/Research_projects/PhiSpace/VignetteData/DC/" # replace this by your own directory where you store ref_dc.qs and query_dc.qs
query <- qread(paste0(dat_dir, "query_Rosa.qs"))
reference <- qread(paste0(dat_dir,"ref_dc.qs"))
query <- subsample(query, key = "celltype", proportion = 0.2, minCellNum = 50, seed = 5202056)To preprocess the data, we apply rank transform to both reference and
query, which is done by PhiSpace::RankTransf. Rank
transform is helpful for removing batch effects in the DC atlas, which
contains hundreds of samples from dozens of studies (Elahi
et al., 2022). In general, normalisation methods should be chosen to
suit individual cases. The only requirement is that both reference and
query are normalised in the same way. In PhiSpace, no
additional harmonisation of reference and query is needed.
# We create a folder called output to store intermediate results to save time
if(!file.exists(paste0(dat_dir, "output/refRanked.qs"))){
query <- RankTransf(query, "counts")
reference <- RankTransf(reference, "data", sparse = F)
qsave(query, paste0(dat_dir, "output/quRanked.qs"))
qsave(reference, paste0(dat_dir, "output/refRanked.qs"))
} else {
query <- qread(paste0(dat_dir, "output/quRanked.qs"))
reference <- qread(paste0(dat_dir, "output/refRanked.qs"))
}PhiSpace Annotation
Some parameters for PhiSpace. phenotypes specifies the
types of phenotypes for PhiSpace to learn. In this particular case, we
care about both cell type and sample source for the query cells, so that
we can predict whether a cell is, say, more in vivo DC-like or in vivo
DC-like. PhiSpaceAssay specifies the assay in reference and
query objects (both SingleCellExperiment or SCE) to use to
compute PhiSpace; in this case rank or the rank normalised
counts. Finally PhiMethod specifies the method to compute
PhiSpace. Currently we support partial least squares (PLS) regression
and principal component regression; the default is PLS which often uses
fewer components for regression and classification tasks (more about
parameter tuning later).
phenotypes <- c("Cell Type", "Sample Source")
PhiSpaceAssay <- "rank"
PhiMethod <- "PLS"There are two key parameters to select for running PhiSpace
annotation: ncomp and nfeat, which controls
the complexity of regression model. We will discuss their selection
towards the end of this article. For now, we use ncomp=30
and load a pre-computed gene
list, containing genes that are most useful for predicting
phenotypes of interest. In our manuscript
why this gene list was well selected.
Now we are ready to apply PhiSpace to continuously annotate the query cells based on bulk reference.
if(!file.exists(paste0(dat_dir, "output/refPhi.qs"))){
c(reference, query) %<-% PhiSpace(
reference,
query,
ncomp = ncomp,
selectedFeat = selectedFeat,
phenotypes = phenotypes,
refAssay = PhiSpaceAssay,
regMethod = PhiMethod,
scale = FALSE,
updateRef = TRUE
)
qsave(reference, paste0(dat_dir, "output/refPhi.qs"))
qsave(query, paste0(dat_dir, "output/quPhi.qs"))
} else {
reference <- qread(paste0(dat_dir, "output/refPhi.qs"))
query <- qread(paste0(dat_dir, "output/quPhi.qs"))
}Visualisation
The most important result from PhiSpace is the predicted phenotype
scores, which we refer to as phenotype space embeddings
of query cells since they characterise the identities (or cell states)
of query cells in a new ‘phenotype space’. The phenotype space
embeddings are stored as a reducedDim object in the query
SCE object. Print it and you will see that this is an cell by phenotype
matrix with each column corresponding to one phenotype defined in the
reference.
head(reducedDim(query, "PhiSpace"))## pDC mono DC1 DC2
## HEF_A2_CGCGTGAGTTTACACG.1 2.677623e-02 -0.4555305 0.1715223 0.199825455
## HEF_A2_GCTCAAATCACCTCAC.1 1.279898e-04 -0.4665929 0.5288902 -0.491670261
## HEF_A2_ATCCGTCCACCCTAAA.1 2.636640e-02 -0.7833520 0.2603889 -0.003032544
## HEF_A2_TTACGTTGTGTATTCG.1 -2.159643e-05 -0.3525349 0.3307356 -0.361853352
## HEF_A2_ACGTACATCATAGGCT.1 5.005486e-02 -0.5999335 0.3023993 -0.044230021
## HEF_A2_GGCAGTCTCATTGCGA.1 6.123582e-02 -0.4776099 0.2922411 -0.116933062
## DC_prec DC MoDC in_vivo
## HEF_A2_CGCGTGAGTTTACACG.1 -0.3048166 -0.27879883 0.4228243 -0.4580049
## HEF_A2_GCTCAAATCACCTCAC.1 -0.5114021 0.71856765 0.2350012 -0.3953980
## HEF_A2_ATCCGTCCACCCTAAA.1 -0.3909576 0.30982959 0.4496005 -0.1379076
## HEF_A2_TTACGTTGTGTATTCG.1 -0.2134217 0.46272537 0.1842853 -0.3770506
## HEF_A2_ACGTACATCATAGGCT.1 -0.4948674 0.38211971 0.2936202 -0.3818141
## HEF_A2_GGCAGTCTCATTGCGA.1 -0.2013902 -0.03738856 0.2896299 -0.1943381
## in_vitro ex_vivo in_vivo_HuMouse
## HEF_A2_CGCGTGAGTTTACACG.1 0.20851264 0.15348864 0.217485671
## HEF_A2_GCTCAAATCACCTCAC.1 0.25279877 0.10363432 0.129166936
## HEF_A2_ATCCGTCCACCCTAAA.1 0.14596210 -0.03699735 0.020866644
## HEF_A2_TTACGTTGTGTATTCG.1 0.17631801 0.39621196 -0.017784582
## HEF_A2_ACGTACATCATAGGCT.1 0.09695693 0.41329455 0.058515516
## HEF_A2_GGCAGTCTCATTGCGA.1 -0.03998713 0.35797421 0.008259488The phenotype space embeddings can be used for various insightful downstream analyses. In the current introductory vignette we focus on some visualisation ideas. Frist we needt to define some colours.
# Customised colour code
DC_cols <- c(
`DC precursor` = "#B3B3B3",DC_prec = "#FFFFB3",
MoDC = "#CCEBC5", cDC1 = "#1B9E77",
cDC2 = "#D95F02",`dendritic cell` = "#B3B3B3",
DC = "#B3DE69", monocyte = "#B3B3B3",
mono = "#80B1D3",`plasmacytoid dendritic cell` = "#7570B3",
DC1 = "#1B9E77",DC2 = "#D95F02",
pDC = "#7570B3",HEF = "#FEE5D9",
Day3 = "#FCAE91",Day6 = "#FB6A4A",
Day9_SP = "#DE2D26", Day9_DP = "#A50F15",
Day9 = "#A50F15",other = "#B3B3B3"
)
DC_cols_source <- c(
ex_vivo = "#B3B3B3",in_vitro = "#A50026",
in_vivo = "#313695",`in_vivo_HuMouse` = "#80B1D3", query = "#B3B3B3"
)Heatmap
The most straightforward visualisation of phenotype space embeddings
is heatmap. Sometimes a heatmap can already provide a lot of insights
for interesting cell states. In PhiSpace package we have
plotPhiSpaceHeatMap, which is a wrapper based on
ComplexHeatmap.
PhiScores_norm <- reducedDim(query, "PhiSpace")
queryLabs <- query$mainTypes
queryLabs[queryLabs %in% c("Day9_SP", "Day9_DP")] <- "Day9"
lvls <- c("DC1", "DC2", "pDC", "HEF", "Day3", "Day6", "Day9")
phenoDict <- list(
cellType = sort(unique(reference$`Cell Type`)),
sampleSource = sort(unique(reference$`Sample Source`))
)
p <- plotPhiSpaceHeatMap(
PhiSpaceScore = PhiScores_norm,
phenoDict = phenoDict,
queryLabs = queryLabs,
queryLvls = lvls,
column_names_rot = 20,
name = "Phenotype space embedding",
row_names_gp = gpar(fontsize = 6),
column_names_gp = gpar(fontsize = 6),
show_row_dend = F,
show_column_dend = T,
# row_title = row_title,
row_title_gp = gpar(fontsize = 6),
column_title_gp = gpar(fontsize = 6, fontface = "bold"),
heatmap_legend_param = list(
title_position = "leftcenter",
title_gp = gpar(fontsize = 6),
grid_height = unit(2, "mm"),
grid_width = unit(2, "mm"),
labels_gp = gpar(fontsize = 5),
legend_direction = "horizontal"
)
) ## `use_raster` is automatically set to TRUE for a matrix with more than
## 2000 rows. You can control `use_raster` argument by explicitly setting
## TRUE/FALSE to it.
##
## Set `ht_opt$message = FALSE` to turn off this message.
# If encounter problem running the following line on Mac, try (re-)install newest version of XQuartz
draw(p, heatmap_legend_side = "top")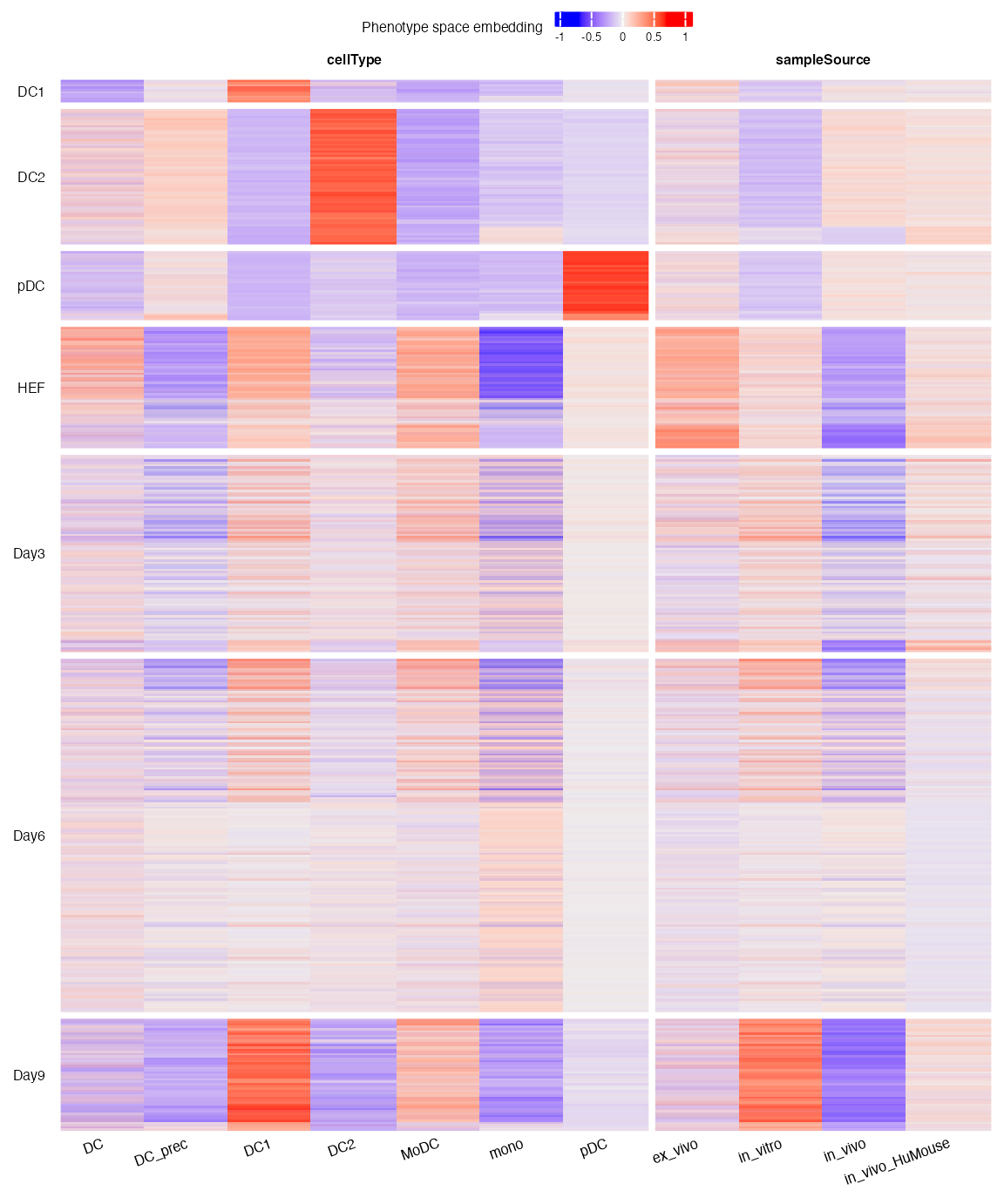
Every column of the heatmap corresponds to a phenotype defined in the bulk reference. Every horizontal line of the heatmap represents a query cell. The query cells are grouped according to their cell types:
- Control cell types: DC1 (type 1 conventional DC), DC2 (type 2 conventional DC) and pDC (plasmacytoid DC). These cell types are in vivo DC subtypes;
- HEF: the starting point of DC reprogramming;
- Day3, Day6, Day9: HEFs after 3, 6 and 9 days of reprogramming.
We can see that the control cell types were predicted to have strong DC1, DC2 and pDC identities. In terms of sample source, they are more in vivo like than in vitro. HEFs have an ambiguous identity since they were not defined in the reference. However, after 9 days of reprogramming, the HEFs were clearly more DC1-like, with strong in vitro identity.
Visualise transitional cell states in phenotype space
The essence of Φ-Space is that we view cell type prediction as dimension reduction. How is it dimension reduction? Look at the heatmap: each cell was represented by the gene expression level of thousands of genes, and now they are represented by 11 dimensions, each measuring their likelihood of belonging to a certain phenotype defined in the reference.
As any other dimension reduction objects, we can use cells’ phenotype space embedding for downstream analyses. One of such analyses is phenotype space PCA, which allows us to visualize both bulk samples and single cells in the same space. The idea is that we first compute a PCA using the phenotype space embeddings of the reference samples, and then project query cells to this PC space via loadings.
queryLabs <- query$mainTypes
queryLabs[queryLabs %in% c("Day9_SP", "Day9_DP")] <- "Day9"
refLabs <- colData(reference)[,"Cell Type"]
YrefHat_norm <- reducedDim(reference, "PhiSpace")
pc_re <- getPC(YrefHat_norm, ncomp = 3)
refEmbedding <- pc_re$scores %>% as.data.frame() %>% mutate(source = reference$`Sample Source`)
# Project query cells via loadings
queryEmbedding <- scale(
PhiScores_norm,
center = T,
scale = F
) %*%
pc_re$loadings %>%
as.data.frame()
plot_ly(
x = ~comp1,
y = ~comp2,
z = ~comp3,
) %>%
add_markers(
data = refEmbedding,
colors = DC_cols,
color = refLabs,
symbol = ~source,
marker = list(
size = 5
)
) %>%
add_markers(
data = queryEmbedding,
color = queryLabs,
marker = list(
symbol = ~"x",
size = 3
)
)Using plotly, we have rendered the PCA results an interactive plot. It is very interesting to observe that, after 9 days of reprogramming, the induced DCs (red crosses) are closer to in vitro rather than in vivo DC1.
PhiSpace parameter tunning
There are two key parameters to select for running PhiSpace annotation:
ncomp: number of PLS (or PCA) components to use for annotation;nfeat: number of features (eg genes, proteins) to use to predict each phenotype (eg cell type, sample source).
PhiSpace uses a regression model to learn phenotypes, such as cell
types, continuously. The parameter ncomp controls the model
complexity: setting ncomp too large (small) may cause
over-(under-)fitting. nfeat controls how many features
PhiSpace uses to make prediction.
PhiSpace provides functions for fully data-driven feature selection based on cross-validation (CV). However, this approach becomes times consuming when the reference sample size becomes large (a common caveat of all CV-based parameter tuning). However, in the current case study where the reference is a bulk atlas, we can still afford to compute CV.
Data-driven parameter tuning (time consuming)
Next we use PhiSpace::tunePhiSpace to do a 5-fold CV
grid search. The function will first plot a loss function plot for
ncomp and then for nfeat. We will see that
ncomp=30 already leads to a very small loss and choosing
larger ncomp doesn’t help further reducing loss. Moreover,
nfeat = 297 seems to minimise the loss and increasing
nfeat will actually increase the loss fucntion.
if(F){ # If you want to run it, change F to T
tuneRes <- tunePhiSpace(
reference, assayName = "rank", phenotypes = phenotypes,
tune_ncomp = TRUE, tune_nfeat = TRUE, ncomp = c(1,50), nfeatLimits = c(10, 5000),
Ncores = 4, Kfolds = 5, seed = 5202056
)
}After running CV tunning, tuneRes$selectedFeat will
return the selected features.
Rule-of-thumb for parameter tunning
As we mentioned, a serious drawback of CV parameter tuning is its
high computational cost. Fortunately PhiSpace is quite robust against
the value of ncomp and a good rule-of-thumb is to set
ncomp to be the number of biologically interesting groups
in the data (ie total number of cell types). In the DC case, setting
ncomp = 11 will lead to a model that is good enough
already. To select nfeat, we can try this simplified
approach:
if(!file.exists(paste0(dat_dir, "output/tuneRes.rds"))){
tuneRes <- tunePhiSpace(
reference, assayName = "rank", phenotypes = phenotypes,
tune_ncomp = F, tune_nfeat = F, ncomp = 11
)
qsave(tuneRes, paste0(dat_dir, "output/tuneRes.rds"))
} else {
tuneRes <- qread(paste0(dat_dir, "output/tuneRes.rds"))
}In this case, no CV tuning actually happened. Instead, we were
telling PhiSpace to stick to ncomp=11 and do not tune
nfeat. What is interesting is
tuneRes$impScores, which gives the importance score for
each gene when used to predict different phenotypes:
head(tuneRes$impScores)## pDC mono DC1 DC2 DC_prec
## F2R 5.051386e-04 2.965319e-04 -0.0005824430 -0.0011207552 -4.280220e-04
## OAZ1 7.861188e-06 4.474383e-06 -0.0001510309 0.0001240407 2.849520e-05
## RPS16 2.117688e-04 -1.503983e-04 -0.0001914275 0.0010778257 -5.906239e-04
## RACK1 3.373127e-05 -2.957098e-05 -0.0001195094 0.0002551319 -4.215025e-05
## ORC6 -7.013646e-04 -5.715517e-04 0.0002952676 0.0038080308 -4.329163e-04
## PDCD7 -1.651123e-04 -4.491403e-04 -0.0006121114 0.0024755905 -1.175561e-04
## DC MoDC in_vivo in_vitro ex_vivo
## F2R 8.656067e-04 4.639430e-04 -7.467888e-05 -2.379497e-03 3.550273e-04
## OAZ1 -1.743889e-06 -1.209670e-05 -1.035714e-04 7.210649e-05 2.649781e-05
## RPS16 -1.775075e-04 -1.796372e-04 3.790844e-04 -1.913091e-04 -3.680781e-05
## RACK1 -4.243059e-05 -5.520199e-05 4.011961e-05 -5.205925e-06 -1.232075e-05
## ORC6 -1.853449e-03 -5.440165e-04 -2.888258e-03 1.928230e-03 7.256920e-04
## PDCD7 -7.067049e-04 -4.249654e-04 -1.199273e-03 2.969786e-04 4.904283e-04
## in_vivo_HuMouse
## F2R 2.099148e-03
## OAZ1 4.967071e-06
## RPS16 -1.509675e-04
## RACK1 -2.259294e-05
## ORC6 2.343365e-04
## PDCD7 4.118664e-04Then we order the importance of features, in descending order, for
each phenotype. Then we can choose the top nfeat features
for each cell type.
nfeat <- 300
selRes <- selectFeat(tuneRes$impScores)
head(selRes$orderedFeatMat)## pDC mono DC1 DC2 DC_prec DC MoDC in_vivo
## [1,] "KRT5" "FCGR3A" "CADM1" "CYYR1" "OLR1" "ALDH1A1" "CCL13" "UQCRH"
## [2,] "TCL1A" "SLC11A1" "CLEC9A" "DBN1" "GLDN" "FCGR3A" "CLDN1" "LYRM2"
## [3,] "PTCRA" "MS4A4A" "ENPP1" "PID1" "ADAM33" "MS4A4A" "MMP12" "EIF1AY"
## [4,] "MAP1A" "ALDH1A1" "PMP22" "IL1R1" "RIMS3" "CCL8" "SUCNR1" "RPL26"
## [5,] "LAMP5" "SLAMF1" "DBN1" "CD1E" "FPR3" "CLC" "C1QA" "HSPB11"
## [6,] "SHD" "NPL" "TACSTD2" "PLPP3" "CAMP" "IDO2" "CCL8" "ATP5F1E"
## in_vitro ex_vivo in_vivo_HuMouse
## [1,] "CCL13" "C1QB" "CDKN3"
## [2,] "CLDN1" "CLDN1" "AURKB"
## [3,] "CD1B" "TFPI" "CTSG"
## [4,] "CD1A" "MMP12" "CKAP2L"
## [5,] "SUCNR1" "ALPK2" "CDC45"
## [6,] "MMP12" "CD1A" "BUB1"
length(unique(as.character(selRes$orderedFeatMat[1:nfeat,])))## [1] 1782For example, we can see that selecting the top 300 features for each
cell type, we end up having 1782 genes selected (due to overlapping
genes), which should be enough for accurate prediction. In practice, we
can adjust the nfeat value till the resulting total number
of feature selected reaches a target number (eg the ‘magic number’ 2000
which is used as default number of genes in Seurat).